Clinical implementation of RNA sequencing in rare genetic disorders
- 22 Apr 2022
- Vicente Yépez
Pathogenic variants in a single gene can lead to rare genetic disorders. Although genetic diagnostics based on sequencing of the patient’s DNA has had a revolutionary impact on rare disease research, around half of the patients remain undiagnosed. This shows that detection and interpretation of potentially pathogenic variants is not always straightforward. In 2017 researchers from the Klinikum Rechts der Isar, TUM, and Helmholtz Zentrum Munich introduced RNA sequencing (RNA-seq) to complement DNA-sequencing in finding the cause for mitochondrial disorders. Since then, several centers worldwide have probed RNA from different tissues and used various approaches to search for the disease-causing mutations and increase diagnostic rates of several types of disorders.
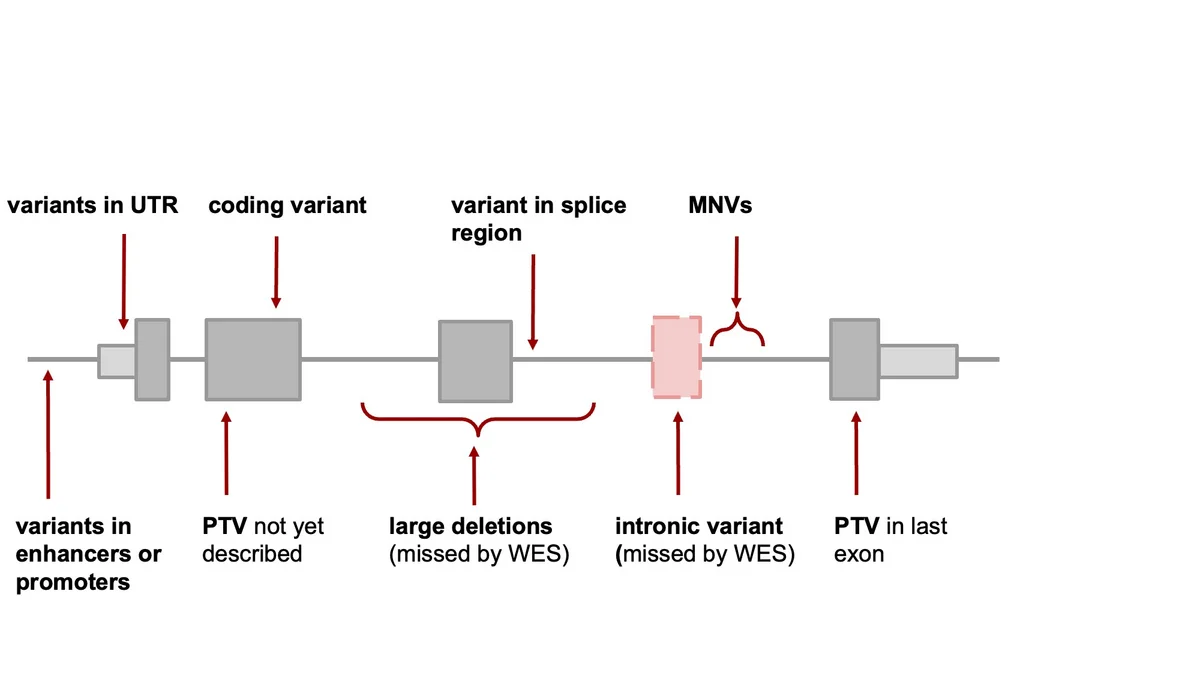
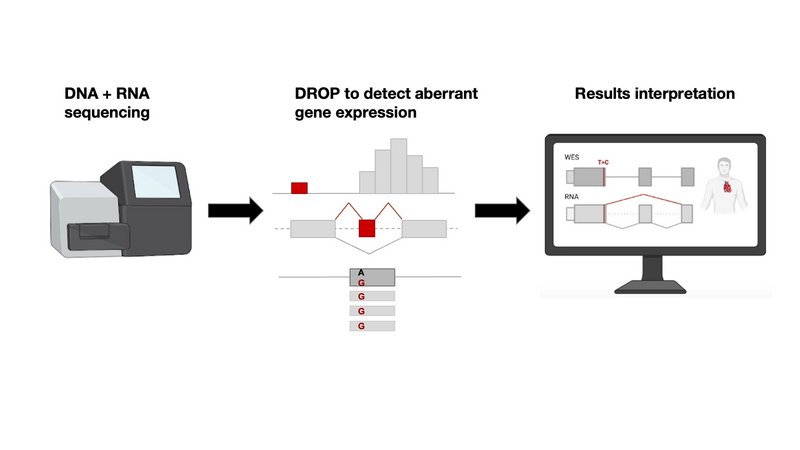
Leveraging on their experience, the same groups have now developed a standard semi-automated workflow from sample probing to gene and variant candidate detection, which in principle can take only 1 week. From the RNA, the researchers are able to statistically detect genes exhibiting aberrant expression, missplicing, and/or expression of only one allele. These so-called ‘outlier’ genes are linked to potentially disease-causing variants and the patient phenotypes to provide a molecular diagnosis. In addition, RNA-seq has the potential to reveal deep intronic variants missed by WES. The researchers gathered one of the largest to-date cohort of whole-exome sequencing (WES) and RNA-seq samples, consisting of 303 patients with suspected genetic disorders, mostly mitochondrial ones and were able to diagnose 32 (16%) of the 205 previously, using WES only, unsolved cases. RNA-seq allowed to capture and validate a broad spectrum of variant types including intronic (often missed in WES), loss-of-function not previously reported, UTRs, splice-region, and synonymous in both recessive and dominant genes.
The researchers also tackled the issue of tissue-specific gene expression, which is one of the biggest limitations of RNA-sequencing, as not every gene is expressed in every tissue. Skin-derived fibroblasts turned out to be the best tissue to probe as more genes related to a variety of diseases are expressed compared to the other clinically used blood and muscle biosamples. Moreover, unlike blood, it is possible to culture fibroblasts for further functional analyses.
RNA-seq is not only useful for pinpointing the causal variants, but can also clarify the molecular pathomechanism, which can help develop possible therapies. The clinical implementation proposed in this study will motivate and accelerate the implementation of RNA-seq in routine rare disease diagnostics.
The authors published and openly shared their automated RNA-seq protocol and computational workflow, DROP, which typically takes less than one week from sample preparation to result reporting. DROP itself was also developed and published by the authors in 2021. DROP is composed of the statistical tools to detect expression outliers ‘OUTRIDER’ and splicing outliers ‘FRASER’. DROP has been used in diagnostics by other research centers worldwide (1,2,3).
This study was led by Vicente Yépez and Mirjana Gusic and the corresponding authors are Julien Gagneur, a GHGA member, and Holger Prokisch. Other GHGA members that contributed are Christian Mertes (set up the robust pipeline), Nicholas Smith (developed the RNA-seq variant calling pipeline), and Thomas Meitinger (supervised). Once operational, GHGA will support such studies by combining large datasets in one secure home, enabling statistical analysis and easy access to reference data sets. Including analysis pipelines developed by partners such as the TUM and Helmholtz Munich into the planned GHGA analysis platform is a further feature planned.
Original Publications:
Yépez, V.A., Gusic, M., Kopajtich, R. et al. Clinical implementation of RNA sequencing for Mendelian disease diagnostics. Genome Med 14, 38 (2022). https://doi.org/10.1186/s13073-022-01019-9