RNA-Sequenzierung unterstützt Diagnose von seltenen genetischen Erkrankungen
- 22 Apr. 2022
- Vicente Yépez
Krankmachende Veränderungen in einem einzigen Gen - genannt Varianten - können zu seltenen genetischen Erkrankungen führen. Gendiagnostik, die auf der Sequenzierung der DNA der Patient:innen beruht, war ein revolutionärer Schritt in der Erforschung seltener Erbkrankheiten. Trotzdem bleibt etwa die Hälfte aller Patient:innen mit einer seltenen genetischen Erkrankung auch heute noch ohne Diagnose. Der Nachweis und die Interpretation potenziell krankheitauslösender Varianten ist nicht immer einfach. 2017 wurde RNA-Sequenzierung als Ergänzung zur DNA-Sequenzierung bei der Diagnose von Patient:innen mit mitochondrialen Erkrankungen eingeführt. Weltweit haben seither mehrere Forschungseinrichtungen und Kliniken RNA aus verschiedenen Geweben untersucht und verschiedene Strategien angewandt, um die krankheitsverursachenden Varianten zu finden und Diagnoseraten für die verschiedenen Arten von Erkrankungen zu erhöhen.
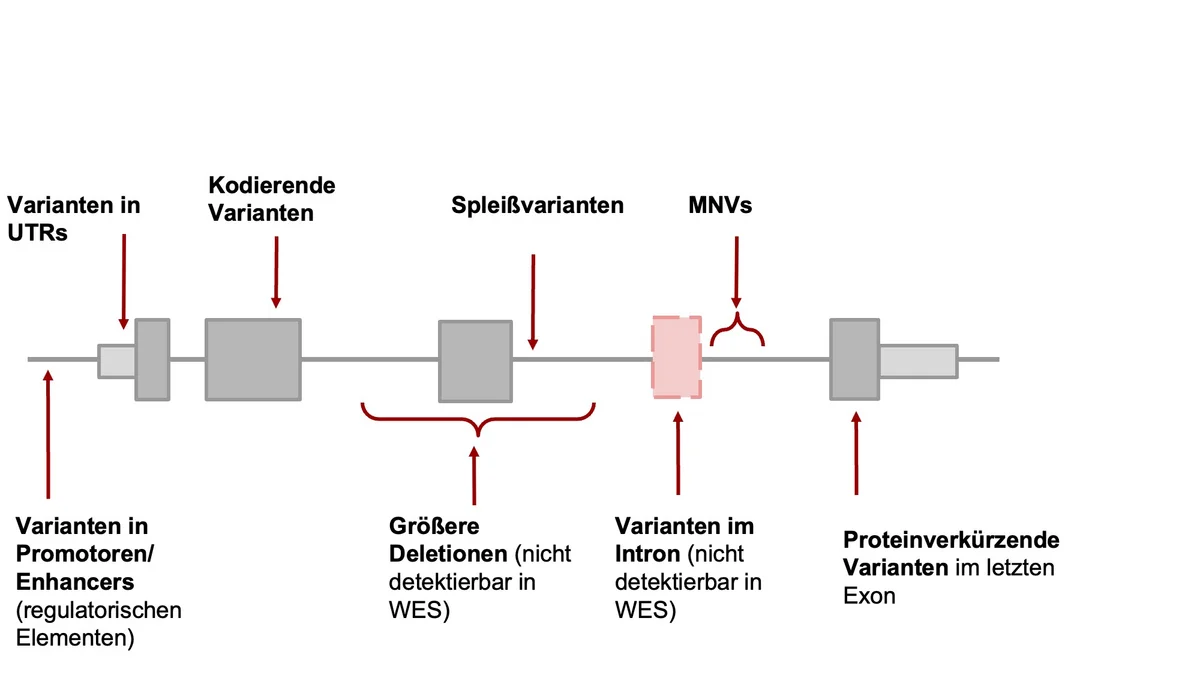
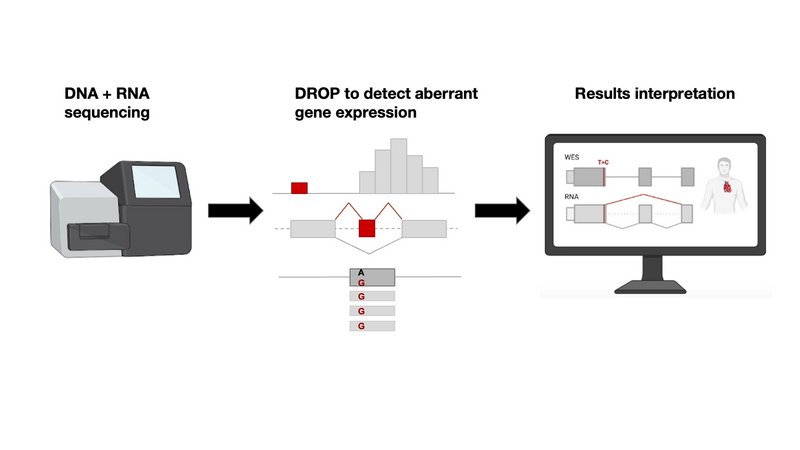
Nun haben Forscher des Klinikums Rechts der Isar, der TUM und des Helmholtz Zentrums München, aufbauend auf ihr Fachwissen, einen standardisierten, halbautomatischen Arbeitsablauf zur Identifizierung von gentischen Varianten entwickelt. Im Prinzip, dauert der Prozess von der Probenentnahme bis zum Nachweis von Genen und Variantenkandidaten nur eine Woche. Mittels statistischer Analysen können aus den RNA Daten Gene aufgespürt werden, die eine ungewöhnlich geringe oder hohe Ableserate (Expression), falsches Spleißen (falsche Entfernung von nicht kodierenden Abschnitten) und/oder limitiertes Ablesen von nur einem Teil der Chromosomenpaare aufweisen. Diese so genannten "Ausreißer"-Gene werden dann mit potenziell krankheitsverursachenden Varianten und den Symptomen der Patient:innen verglichen, um eine genetische Diagnose zu stellen. Darüber hinaus hat RNA-seq das Potenzial Varianten aufzudecken, die von WES übersehen werden. Die Forscher haben, Stand heute, dafür eine der größten Patientenkohorte basierend auf Whole Exome Sequencing (Sequenzierung der DNA) und RNA-Seq Proben zusammen getragen. Die Proben stammen von 303 Patienten mit vermuteten genetischen Störungen. 32 (16 %) der 205 Patienten, zuvor nach alleiniger WES Diagnose ungeklärten Krankheiten, konnten mit dem neuen Protokol diagnostiziert werden. Da WES nur die Protein-kodierenden Exone analysiert, können Varianten in den DNA Bereichen dazwischen, den Intros, nicht über WES identifiziert werden. RNA-seq ermöglicht folglich die Erfassung und Validierung eines breiten Spektrums von Varianten: darunter intronische Varianten, Funktionsverluste im letzten Exon, UTRs, Spleißregionen und synonyme Varianten sowohl in rezessiven als auch in dominanten Genen.
Die Forscher befassten sich auch mit der Frage der gewebespezifischen Genexpression. Da nicht jedes Gen in jedem Gewebe exprimiert wird, ist Gewebespezifität eine der größten Einschränkungen der RNA-Sequenzierung. Aus der Haut stammende Fibroblasten erwiesen sich als das geeignetste Gewebe für die Untersuchung. Sie exprimieren mehr Gene, die mit einer Vielzahl von Krankheiten in Zusammenhang stehen, als die anderen in der Klinik erprobten Blut- oder Muskelgewebe. Außerdem ist es, im Gegensatz zu Blut möglich, Fibroblasten für weitere funktionelle Analysen zu kultivieren.
RNA-seq ist nicht nur geeignet, die ursächlichen Varianten zu ermitteln, sondern kann auch zur Klärung des molekularen Pathomechanismus und damit zur Entwicklung möglicher Therapien beitragen. Die vorliegende Studie und die darin erprobte und automatisierte klinische Anwendung von RNA-seq wird die Einführung der Methode in der Routinediagnostik seltener Erbkrankheiten fördern und hoffentlich beschleunigen.
Die Autoren haben ihr automatisiertes RNA-seq-Protokoll und den zugehörigen bioinformatischen Workflow, DROP, in einer Fachzeitschrift veröffentlicht und frei zugänglich auf Github gemacht. In der Praxis dauert der Ablauf von der Probenvorbereitung bis zur zum Ergebnisse in der Regel weniger als eine Woche. DROP selber wurde ebenfalls von den Autoren entwickelt und im Jahr 2021 veröffentlicht. DROP besteht aus den statistischen Tools zur Erkennung von Expressionsausreißern "OUTRIDER" und Spleißausreißern "FRASER". DROP wurde bereits von anderen Forschungszentren weltweit in der Diagnostik eingesetzt (1,2,3).
Diese Studie wurde von Vicente Yépez und Mirjana Gusic geleitet und die Korrespondenzautoren sind Julien Gagneur, ein GHGA Mitglied, und Holger Prokisch. Weitere GHGA-Mitglieder, die einen Beitrag leisteten, sind Christian Mertes (Einrichtung der robusten Pipeline), Nicholas Smith (Entwicklung der RNA-seq-Pipeline zur Variantenerkennung) und Thomas Meitinger (Supervision). Sobald GHGA einsatzbereit ist, wird es derartige Studien unterstützen, indem es große Datensätze an einem sicheren Ort zusammenführt, so statistische Analysen ermöglicht und den Zugang zu Referenzdatensätzen erleichtert. Die Einbeziehung von Analysepipelines, die von Partnern wie der TUM und Helmholtz München entwickelt wurden, in die geplante GHGA-Analyseplattform ist ein weiteres geplantes Feature.
Original-Publikationen:
Yépez, V.A., Gusic, M., Kopajtich, R. et al. Clinical implementation of RNA sequencing for Mendelian disease diagnostics. Genome Med 14, 38 (2022). https://doi.org/10.1186/s13073-022-01019-9