



Veröffentlichungen von bioinformatischen Workflows mit GHGA-Beteiligung
- 29 Juli 2022
- Nicholas Smith
Das Bioinformatik-Team von GHGA macht stetig Fortschritte in seinen Bemühungen standardisierte, vergleichbare und reproduzierbare bioinformatische Workflows für die Forschungsgemeinschaft bereitzustellen. Zuletzt war GHGA an der Veröffentlichung von drei bioinformatischen Workflows (sarek 3.0, nanoseq 3.0 und DROP 1.2) beteiligt, die wir - neben den laufenden Bemühungen - hier vorstellen.
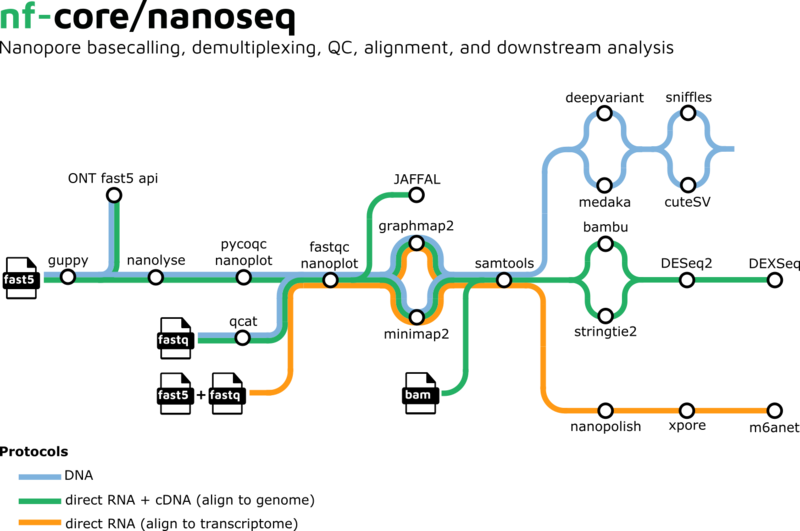
In Zusammenarbeit mit den nf-core-Entwicklern haben die Bioinformatiker von GHGA an der Weiterentwicklung von sarek 3.0 gearbeitet! Bei der neuen Version handelt es sich um einen komplett modularen Workflow. Er ist unabhängig von der Rechnerumgebung und enthält dutzende Community-Tools, die Keimbahn- und Tumor-Variantenerkennung von Anfang bis Ende ermöglichen. Das Team hat auch mit den nf-core-Entwicklern von der sogenannten Long-Read Sequenziertechnologie zusammengearbeitet, um den Workflow nanoseq zu verbessern und zu erweitern. Die neue Hauptversion nanoseq 3.0 enthält jetzt neben RNA-Modifikation und Fusionserkennung auch DNA Varianten Detektion. Die neue Funktionalitäten sind im bytesize Video zusammengefasst
Außerdem entstand in Zusammenarbeit mit dem Gagneur Labor an der TUM eine neue Version der Detection of RNA Outlier Pipeline (DROP 1.2). DROP, das bereits von dem europäischen Solve-RD Programm benutzt wird, konzentriert sich auf die Erkennung seltener transkriptomischer Ereignisse während fehlerhafter Spleißprozesse oder Regulation der Geneexpresion inklusive der Detektion von Expression von nur einem Allel - der sogenannte monoallelischen Expression. Über eine frühere Veröffentlichung und eine Publikation im April haben wir hier berichtet.
Neben diesen Meilensteinen arbeitet der GHGA Workflow Workstream derzeit mit den Entwicklern von nf-core Workflow scrnaseq 2.0 und scatac zusammen, um standartisierte, qualitativ hochwertige und reproduzierbare Workflows für Einzelzell-RNA-seq- und ATAC-seq-Datensätze zu entwickeln. GHGA ist auch an den Benchmarking-Bemühungen des deutschen Next Generation Sequencing Competence Network (NGS-CN) beteiligt, um den Benchmarking-Prozess für Sequenzierzentren in ganz Deutschland zu standardisieren.
Wir freuen uns darauf, weiterhin nützliche Lösungen zu entwickeln, und schätzen das Mitwirken der Community! Bitte füllen Sie die Umfrage aus, um die Produkte und Workflows, die GHGA für die Nutzer entwickelt und bereitstellt, mitzugestalten: https://www.ghga.de/news/detail/ghga-survey-for-community-driven-development
Offenen Zugang zu allen Workflows finden Sie auf Github :
https://github.com/nf-core/sarek
https://github.com/nf-core/nanoseq